








Dr James Udelson is the Chief of the Division of Cardiology as well as the Director of Nuclear Cardiology at Tufts Medical Center. Dr Udelson’s research interests involve new therapeutic modalities in the setting of heart failure as well as chronic coronary artery disease.
Dr Udelson began his presentation by highlighting the problems of clinical trials enrolling heart failure (HF) patients. Clinical trials are extremely expensive and the majority of patients do not contribute to the primary endpoint. This is demonstrated in contemporary clinical trials in HF, where event rates are usually low. In the recent Prospective Comparison of Angiotensin Receptor–Neprilysin Inhibitor with Angiotensin-Converting–Enzyme Inhibitor to Determine Impact on Global Mortality and Morbidity in Heart Failure (PARADIGM-HF) trial, the 1-year rate of cardiovascular death/hospitalisation was 10–12 %; therefore, in order to demonstrate statistical significance, the trial needed to enrol >8,000 patients.1 In the 2012 Abciximab Intracoronary versus intravenous Drug Application in ST-elevation myocardial infarction (AIDA STEMI) trials, a three-point composite endpoint was used, but despite recruiting >2,000 patients, the trial had an event rate of only 7 % at 90 days and statistical significance was not achieved.2
As the use of cardiac unloading moves into less severely ill patients, such low event rates will become an issue in trial design. There is therefore much interest in biomarkers as surrogates in trials. A marker is considered a surrogate when it is in the causal path between the remedy and the outcome.3,4 Markers may be serum biomarkers, such as troponins or natriuretic peptides, or imaging biomarkers, such infarct size, left ventricle volume and ejection fraction. All intervention effects pass through the marker in the causal path or are captured by the marker.
Dr Udelson focused on the use of infarct size measured by cardiac magnetic resonance (CMR) imaging as a plausible marker for myocardial infarction (MI). A large body of data shows that infarct size influences established clinical outcomes such cardiovascular death and HF hospitalisation. However, therapeutic interventioninduced changes in the surrogate marker need to be reflected in changes in the clinical outcome. At present, no marker is able to achieve this standard. Biomarkers that are ‘prognostic’ are not necessarily good surrogate markers in terms of assessing the effects of therapy. As an example, premature ventricular complexes following MI are strongly associated with an unfavourable prognosis. However, in the Cardiac Arrhythmia Suppression Trial (CAST), suppression of premature ventricular complexes led to an increased mortality.5 Likewise, low high-density lipoprotein is prognostic of an increased risk of incident coronary artery disease, but the use of the cholesteryl ester transfer protein inhibitor torcetrapib to raise high-density lipoprotein levels increased mortality due to its effects on glucose, insulin and HbA1c in subjects in the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE) trial.6
The question of whether infarct size is an appropriate surrogate endpoint was addressed in a recent study by Dr Udelson’s research team: a pooled patient-level meta-analysis from 10 randomised primary percutaneous coronary intervention in ST-elevation MI trials (n=2,632 patients) in which infarct size was assessed within 1 month after randomisation by either CMR imaging or technetium-99m single-photon emission CT, with clinical follow-up for ≥6 months. A strong correlation was seen between infarct size (per 5 % increase) and subsequent mortality (1.19; 95 % CI [1.18 to 1.20]; p<0.0001) and hospitalisation for HF (adjusted hazard ratio: 1.20; 95 % CI [1.19–1.21]; p<0.0001), independent of age, sex, diabetes, hypertension, hyperlipidaemia, current smoking, left anterior descending versus non-left anterior descending infarct vessel, symptom-to-first device time and baseline thrombolysis in MI flow 0/1 versus 2/3. Infarct size was not significantly related to subsequent reinfarction. For every 1 % reduction in infarct size, there was a 16 % reduction in HF hospitalisation but no effect on all cause mortality.7 The investigators plan to meet with the US Food and Drug Administration (FDA) to discuss whether these data support the incorporation of infarct size into trial outcomes.
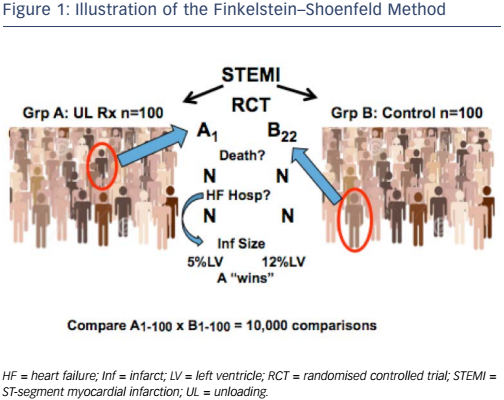
If the FDA approves the use of infarct size as a surrogate endpoint in clinical trials, the next challenge will be how to incorporate it. Two methods may be useful: the Finkelstein–Schoenfeld method8 and the ‘win ratio’,9 which involve a hierarchical comparison of events/timing in pairs of patients from the groups in the trials. The analyses account for clinical priority (e.g. death is more important than HF hospitalisation) and allow the potential incorporation of longitudinal measures such as the change in 6-minute walk distance or biomarkers. Most importantly, these methods enable all patients in a trial to contribute to the endpoint.
Dr Udelson illustrated this concept by considering a hypothetical randomised controlled ST-elevation MI trial that investigates cardiac unloading. Group A comprises 100 patients who receive unloading, while group 2 comprises 100 controls (see Figure 1). The investigator takes a patient (e.g. patient 1) from group A and another (e.g. patient 22) from group B and compares them. At the first level of hierarchy, the investigator compares whether either patient died. If B22 died but A1 was alive at study completion, then A ‘wins’ that comparison. If both patients died but A1 died at 12 months while B22 died at 8 months, then group A ‘wins’. However, in HF or acute MI trials, most patients do not die. The following step is to consider the next level, i.e. HF hospitalisations. If B22 was hospitalised but A1 was not, then group A ‘wins’. In ST-elevation MI, neither of these events may occur so it may be necessary to move to a marker with a plausible relationship with outcomes. If A1 has an infarct size of 5 % but B22 has an infarct size of 12 %, then group A 'wins' that comparison. If each person in group A is compared in this way with each person in group B, we obtain 10,000 comparisons.
This approach is familiar to the FDA. It was used in cohort B of the transcatheter aortic valve replacement group in the Placement of AoRtic TraNscathetER Valves (PARTNER) trial, which had co-primary endpoints of all-cause mortality (p<0.0001 favouring the device) and a hierarchical composite of death/recurrent hospitalisation, analysed by the Finkelstein–Schoenfeld method. Results showed superiority of the device (p<0.0001).10
Dr Udelson concluded that incorporating validated markers into hierarchical composites may allow reasonable sample sizes for trials of approaches to ST-elevated MI such as mechanical circulatory support.